 By Brad Dieter, MS, CSCS, CISSN. I just returned from the American College of Sports Medicine conference in which the theme was “Exercise is Medicine”. I would argue that exercise is indeed medicine and that diet and exercise are the most powerful “medicines” to combat the current epidemic of modern chronic diseases such as obesity, type 2 diabetes, and heart disease. In fact, Robert H. Butler stated, “If exercise could be purchased in a pill, it would be the single most widely prescribed and beneficial medicine in the nation”. The advances of modern science have reached a point where they can mimic the effects of exercise through pharmacological agents at a cellular level. The purpose of this article is to review some of these pharmacological agents and provide a comparison of them to actual real exercise in regards to obesity, type 2 diabetes, and heart disease. Unfortunately, in order to fully grasp the concept of “exercise in a pill” I have to explain some rather technical concepts.
By Brad Dieter, MS, CSCS, CISSN. I just returned from the American College of Sports Medicine conference in which the theme was “Exercise is Medicine”. I would argue that exercise is indeed medicine and that diet and exercise are the most powerful “medicines” to combat the current epidemic of modern chronic diseases such as obesity, type 2 diabetes, and heart disease. In fact, Robert H. Butler stated, “If exercise could be purchased in a pill, it would be the single most widely prescribed and beneficial medicine in the nation”. The advances of modern science have reached a point where they can mimic the effects of exercise through pharmacological agents at a cellular level. The purpose of this article is to review some of these pharmacological agents and provide a comparison of them to actual real exercise in regards to obesity, type 2 diabetes, and heart disease. Unfortunately, in order to fully grasp the concept of “exercise in a pill” I have to explain some rather technical concepts.
Obesity, Type 2 Diabetes, and Heart Disease
It is quite clear that obesity, type 2 diabetes, and heart disease are the biggest health issues in America. While this is an oversimplification, I believe these three diseases are connected and are related to metabolic dysfunction. Additionally, I argue that this metabolic dysfunction can be attributed to lipid accumulation and mitochondrial dysfunction for when we examine the data regarding of these three diseases, we observe both lipid accumulation and mitochondrial dysfunction.
Exercise and Muscle Adaptation
Exercise induces physiological changes in every system of our body with our muscle tissue undergoing the most dramatic and metabolically significant changes. The adaptations in muscle that arise from exercise are both acute and chronic, with each type impacting metabolic processes and function. The most salient adaptations to this discussion include non-insulin dependent glucose transport, increased intramuscular fatty-acid oxidation, increased insulin sensitivity, and enhanced mitochondrial function. Furthermore, each of these adaptations aid in attenuating and reversing lipid accumulation and mitochondrial dysfunction. There are currently several hypotheses on the pathogenesis of these diseased states with lipid accumulation and mitochondrial dysfunction serving as a uniting factor in each of them. Taken together, this suggests the cellular adaptations to muscle tissue that arise from exercise are beneficial to obesity, type 2 diabetes, and heart disease. Therefore, inducing these adaptations or activating the same signaling pathways through pharmacological interventions may produce benefits similar to exercise. Two main signaling processes involved in exercise that are currently being targeted with pharmacological agents are AMPK and the PGC1/PPAR co-activator/transcription factor.
AMPK
One of the best understood metabolic enzymes is AMP-activated protein kinase (AMPK), a master regulator of cellular and organism metabolism whose function is conserved in all eukaryotes (1). AMPK is involved in glucose homeostasis and is direct impacted through exercise. Briefly, AMPK is activated in response to an increase in the AMP/ATP ratio which increases during exercise with ATP is utilized for fuel (See Figure 1). This indicates that during exercise, AMPK signals GLUT4 translocation and non-insulin dependent glucose disposal.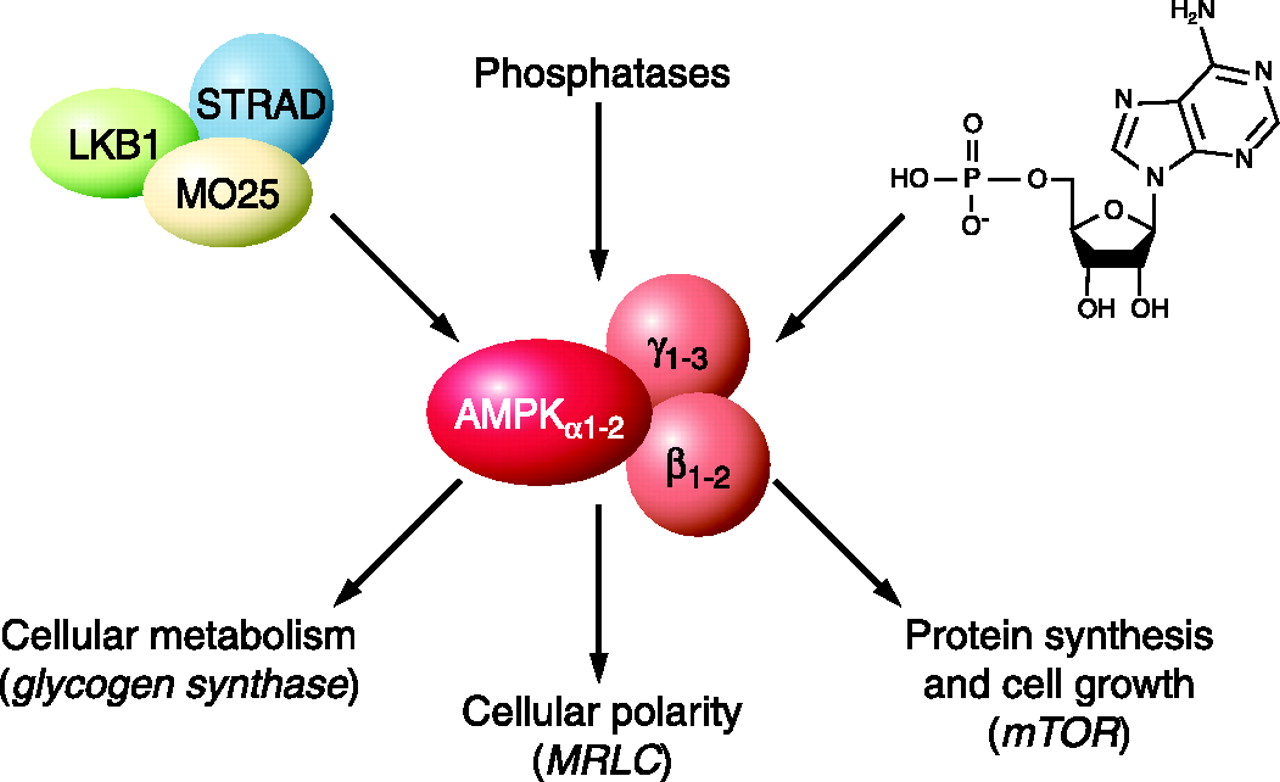
A brilliant paper by Fryer et al. (2002) demonstrated all of the necessary components to indicate the critical role AMPK plays in the beneficial adaptations of exercise in glucose homeostasis and how pharmacological therapy can mimic cellular conditions present during exercise (2). In the paper, the authors successfully demonstrated that AMPK does indeed stimulate glucose uptake into cells. More importantly, they demonstrated that pharmacological agents can mimic the effect of exercise by “activating” AMPK. In their study they demonstrated that AICAR, a synthetic analogue of AMP, effectively increased AMPK signaling and glucose transport in skeletal muscle tissue. These results are not unique, another study in 2002 demonstrated that AICAR increased glucose transport (3). Put simply, AICAR mimics muscle contraction and AMPK activation by increasing the AMP/ATP ratio (Figure 2). Furthermore, it has been shown that the anti-diabetic drugs Rosiglitazone and Metformin improve glucose control through the AMPK pathway (4). Taken together, this indicates that AICAR, and other pharmacological agents, can mimic the acute effects exercise has on glucose disposal through activating AMPK and GLUT4 translocation.
PPAR- γ and PGC-1 Agonists
Exercise can induce adaptations in genetic expression in muscle tissue which result in the manufacturing of certain proteins that alter muscle cell function. Many of these adaptations occur via Peroxisome proliferator-activated receptors (PPAR) and its co-activator PGC-1. Briefly, PPAR is a nuclear receptor that regulates glucose and fatty acid metabolism and PGC-1 effects biologic responses that equip the cell to meet the energy demands of a changing environment, including augmentation of mitochondrial biogenesis, cellular respiration rates, and energy substrate uptake and utilization (5). While we can discuss PGC-1 and PPAR-γ separate from AMPK, they can also be discussed together as AMPK influences PGC-1 and PPAR (6, 7). This discussion is interesting but is outside the scope of this article. A brief explanation is depicted in figure 3, and while there are additional AMPK independent pathways (i.e. p38 MAPK) I feel this figure shows the concept easily enough.
Put simply, type 2 diabetes is a metabolic disease in which we observe dysfunction in the metabolism of both carbohydrate and fat. As metabolism is initially controlled at a cellular level we observed differences in gene expression and nuclear receptor activation between type 2 diabetics and normal, healthy individuals. One key difference is a lower expression of PGC1-α in type 2 diabetics (8). This down of PGC1-α is concurrent with decreased oxidative phosphorylation, indicating mitochondrial dysfunction (9). Exercise has been shown to upregulate transcription of PGC-1. This upregulation leads to enhanced lipid metabolism, improved mitochondrial function, and mitochondrial biogenesis (10, 11). Taken together, this indicates that exercise improves lipid accumulation and mitochondrial function through PGC-1α
PGC-1 agonist drugs have been shown to mimic the cellular adaptations of exercise through PGC-1 and that the adaptations are functional as enhance exercise endurance in rats (12)
While it is clear that PGC-1 is a key player in exercise-induced skeletal muscle adaptations, specifically mitochondrial biogenesis, it is not the only player. Researchers have demonstrated that there are other mechanisms that contribute to this exercise induced adaptations with PGC-1α knockout mice (13). This appears to indicate that while PGC-1 agonists can have a beneficial effect on skeletal muscle, there are other mechanisms through which exercise induces beneficial adaptions. Taken together, I would conclude that while these drug therapies may be beneficial, the specific targets of these drugs (PGC-1 agonists) do not mimic the full extent of exercise as I will touch on next.
Both PPAR-γ and its co-activator, PGC-1, have been targets for pharmacological therapy to treat type 2 diabetes, specifically thiazolidinediones (TZD’s) (14). These agents have shown to be efficacious for glucose control in diabetes even though the mechanisms were not fully explained at the time. Additionally, PPAR-γ agonists do not specifically target the muscle cells, they also interact with adipocytes and promote lipid storage (Figure 4). Taken together, it appears these pharmacological agents may mimic exercise and are effective in improving lipid metabolism (PPAR-γ) and improving mitochondrial function and biogenesis (PGC-1) in muscle cells; however, they are not muscle cell specific, indicating they may also increase body fat which has negative consequences on inflammation and metabolism.
In Summary
As evidenced in this article, pharmacological therapy can mimic many of the effects of exercise at a cellular level and may indeed have a place in treating obesity. These therapies may be powerful tools in the initial treatment to help “kick start” an e xercise regimen and begin to shift our metabolic engine in a better direction. That being said, I do not believe we have truly replaced exercise. As witnessed with these drugs, many of their positive effects at the muscle tissue may be counteracted in the adipose tissue (i.e. TZDs). Furthermore, they do not confer the same benefits in regards to psychological health or skeletal health. As with all drugs they ought to be a tool to augment the role of a healthy lifestyle that includes proper diet and exercise.
xercise regimen and begin to shift our metabolic engine in a better direction. That being said, I do not believe we have truly replaced exercise. As witnessed with these drugs, many of their positive effects at the muscle tissue may be counteracted in the adipose tissue (i.e. TZDs). Furthermore, they do not confer the same benefits in regards to psychological health or skeletal health. As with all drugs they ought to be a tool to augment the role of a healthy lifestyle that includes proper diet and exercise.
BIO – Brad is a Ph.D. student at the University of Idaho. He is in a joint program studying exercise physiology and ethics. He received his M.S. degree in biomechanics and has a passion for evolutionary nutrition and the potential it has to combat the current declining state of health. Brad has a background as an athlete, basketball coach, and strength and conditioning coach. He loves learning about the human body, how it functions, and how we can optimally nourish it. He is focusing his doctoral work on how diet and exercise influence metabolism and health in relation to chronic, lifestyle diseases such as diabetes and heart disease.


